What is biliary atresia (ba)?
Biliary atresia is a rare disease of the liver and bile ducts that occurs in infants. Symptoms of the disease appear or develop about two to eight weeks after birth.
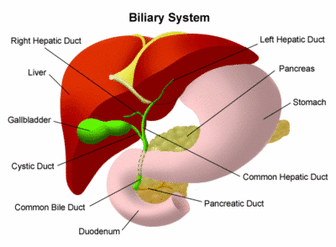
Cells within the liver produce liquid called bile. Bile helps to digest fat. It also carries waste products from the liver to the intestines for excretion.
This network of channels and ducts is called the biliary system. When the biliary system is working the way it should, it lets the bile drain from the liver into the intestines.
When a baby has biliary atresia, bile flow from the liver to the gallbladder is blocked. This causes the bile to be trapped inside the liver, quickly causing damage and scarring of the liver cells (cirrhosis), and finally liver failure.
About one in 15,000 to 20,000 babies do not have complete bile ducts. Biliary atresia seems to affect girls more than boys. Within the same family, it is common for only one child in a pair of twins or only one child within the same family to have it. Asians and African-Americans are affected more frequently than Caucasians.
Babies with biliary atresia usually appear healthy when they are born. Symptoms of the disease typically appear within the first two weeks to two months of life. Those symptoms include:
Jaundice may be present with other liver disorders, so several tests are needed to get the correct diagnosis.
Kasai Procedure
A Kasai procedure or hepatoportoenterostomy is done . The Kasai procedure is an operation to create an open duct so bile can drain from the liver. It is named after the surgeon who developed it.
The surgeon removes the damaged ducts outside of the liver (extrahepatic ducts) and replaces them with a piece of the baby's own intestine. This new duct allows bile to pass from the liver into the intestine.
After this procedure, infants are usually in the hospital for 7 to 10 days to heal. Medications are used to prevent excessive fluid build-up in the abdomen (ascites). Long-term antibiotic therapy is also given to reduce the risk of infection.
With an experienced surgeon, the Kasai procedure is successful in 60 to 85 percent of the patients. This means that bile drains from the liver and the jaundice goes down.
The Kasai procedure is not a cure for biliary atresia, but it does allow babies to grow and have fairly good health for several years.
When this procedure does not work, it is usually because the blocked bile ducts are inside the liver (intrahepatic), as well as outside the liver (extrahepatic). If this is the case, liver transplantation can correct this problem.
Success with this procedure is related to:
Long-term survival after the Kasai procedure is affected by the presence of progressive liver disease (cirrhosis) and the development of portal hypertension (high blood pressure in the portal vein that carries blood to the liver).
Nearly one-half of all infants who have had a Kasai procedure require liver transplantation before age 5. Older children may continue to have good bile drainage and no jaundice.
Some children may develop portal hypertension and have gastrointestinal bleeding, accumulation of fluid in the abdomen (ascites) and overactivity of the spleen (hypersplenism).
Eighty-five percent of all children who have biliary atresia will need to have a liver transplant before they are 20 years old. The remaining 15 percent have some degree of liver disease. Their disease can be managed without having a transplant.
Liver Transplant
If there is still not enough bile flow with the Kasai procedure, liver transplantation is a final option. A liver transplant operation removes the damaged liver and replaces it with a new liver from a donor.
After transplant surgery, the child's health may improve quite quickly. However, the child's body might reject the new organ. To prevent rejection, a strict schedule of anti-rejection medications must be taken.
After a transplant, ongoing lifelong care is required. Frequent contact with physicians and other members of the transplant team is also necessary.
Cells within the liver produce liquid called bile. Bile helps to digest fat. It also carries waste products from the liver to the intestines for excretion.
This network of channels and ducts is called the biliary system. When the biliary system is working the way it should, it lets the bile drain from the liver into the intestines.
When a baby has biliary atresia, bile flow from the liver to the gallbladder is blocked. This causes the bile to be trapped inside the liver, quickly causing damage and scarring of the liver cells (cirrhosis), and finally liver failure.
About one in 15,000 to 20,000 babies do not have complete bile ducts. Biliary atresia seems to affect girls more than boys. Within the same family, it is common for only one child in a pair of twins or only one child within the same family to have it. Asians and African-Americans are affected more frequently than Caucasians.
Babies with biliary atresia usually appear healthy when they are born. Symptoms of the disease typically appear within the first two weeks to two months of life. Those symptoms include:
- Jaundice- yellow coloring of the skin and eyes due to a very high level of bilirubin (bile pigment) in the bloodstream.
Jaundice caused by an immature liver is common in newborns. It usually goes away within the first week to 10 days of life. A baby with biliary atresia usually appears normal at birth, but develops jaundice at two or three weeks after birth. - Dark urine- caused by the build-up of bilirubin (a breakdown product from hemoglobin) in the blood. The bilirubin is then filtered by the kidney and removed in the urine.
- Acholic stools (clay-colored stools)- because no bile or bilirubin coloring is being emptied into the intestine. Also, the abdomen may become swollen from a firm, enlarged liver.
- Weight loss and irritability- develop when the level of jaundice increases.
Jaundice may be present with other liver disorders, so several tests are needed to get the correct diagnosis.
- Blood tests are done to tell if there are liver function abnormalities. They can also tell the cause (etiology) of jaundice.
- X-rays of the abdomen look for an enlarged liver and spleen.
- Abdominal ultrasound can tell if there is a small gall bladder or none at all.
- Nuclear test, called an HIDA scan, determines the flow of bile. In this scan, a radioactive dye is injected into the infant's vein.
The dye acts like bilirubin. If the baby has biliary atresia, the liver will take up the dye but it will not be able to flow through the damaged biliary system into the small intestine. - Liver Biopsy tells if an infant is likely to have biliary atresia. In a liver biopsy, a tiny sample of the liver is removed with a needle. That sample is then looked at under a microscope.
A liver biopsy is very reliable. If the biopsy shows that the infant probably has biliary atresia, further surgery will confirm the diagnosis and treat the condition. - Diagnostic surgery confirms if an infant has biliary atresia. Surgery allows doctors to see if there is an injured piece of the bile ducts going from the liver to the intestine. This could prevent normal bile flow from the liver.
- Operative cholangiogram is done when a gall bladder is present. At operation, a dye is injected through the gall bladder and goes through the bile ducts. An X-ray is done to learn if the dye flows normally into the intestine and the liver. In infants with biliary atresia, the dye does not usually flow out of the gall bladder due to the blocked ducts.If the ducts are normal or open (patent) and the dye flows the way it should, biliary atresia is ruled out. A bigger liver biopsy (tissue sample) is then done to find the cause of the liver disorder.
Biliary atresia is diagnosed after the ducts have been checked and when the cholangiogram shows that the bile ducts are not open. Then infants usually undergo an operation called the Kasai procedure.
Kasai Procedure
A Kasai procedure or hepatoportoenterostomy is done . The Kasai procedure is an operation to create an open duct so bile can drain from the liver. It is named after the surgeon who developed it.
The surgeon removes the damaged ducts outside of the liver (extrahepatic ducts) and replaces them with a piece of the baby's own intestine. This new duct allows bile to pass from the liver into the intestine.
After this procedure, infants are usually in the hospital for 7 to 10 days to heal. Medications are used to prevent excessive fluid build-up in the abdomen (ascites). Long-term antibiotic therapy is also given to reduce the risk of infection.
With an experienced surgeon, the Kasai procedure is successful in 60 to 85 percent of the patients. This means that bile drains from the liver and the jaundice goes down.
The Kasai procedure is not a cure for biliary atresia, but it does allow babies to grow and have fairly good health for several years.
When this procedure does not work, it is usually because the blocked bile ducts are inside the liver (intrahepatic), as well as outside the liver (extrahepatic). If this is the case, liver transplantation can correct this problem.
Success with this procedure is related to:
- Age. Surgery is most successful in infants younger than two to three months of age.
- Extent of liver damage (cirrhosis) at the time of surgery.
- The number and size of microscopic ducts in the scarred tissue that can drain bile.
- The experience of the surgical and medical team. Centers with teams made up of specialists with extensive experience have success rates that are greater than those centers with less experienced teams.
Long-term survival after the Kasai procedure is affected by the presence of progressive liver disease (cirrhosis) and the development of portal hypertension (high blood pressure in the portal vein that carries blood to the liver).
Nearly one-half of all infants who have had a Kasai procedure require liver transplantation before age 5. Older children may continue to have good bile drainage and no jaundice.
Some children may develop portal hypertension and have gastrointestinal bleeding, accumulation of fluid in the abdomen (ascites) and overactivity of the spleen (hypersplenism).
Eighty-five percent of all children who have biliary atresia will need to have a liver transplant before they are 20 years old. The remaining 15 percent have some degree of liver disease. Their disease can be managed without having a transplant.
Liver Transplant
If there is still not enough bile flow with the Kasai procedure, liver transplantation is a final option. A liver transplant operation removes the damaged liver and replaces it with a new liver from a donor.
After transplant surgery, the child's health may improve quite quickly. However, the child's body might reject the new organ. To prevent rejection, a strict schedule of anti-rejection medications must be taken.
After a transplant, ongoing lifelong care is required. Frequent contact with physicians and other members of the transplant team is also necessary.